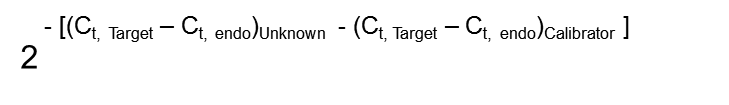实时定量PCR原理和操作
1. 实时定量PCR:绝对定量法
传统PCR是通过电泳检查PCR扩增后的DNA终点产物;而定量PCR 是利用荧光检测器实时监控每个PCR循环后产生荧光的强弱,在检测器能最早检测荧光的PCR循环数被记录为阈值循环数。这一值与DNA模板的拷贝数的Log值呈负线性相关性,即原始DNA样品中的底物模板拷贝数越高,荧光能越早被检测到,阈值循环数也越小。在SYBR Green做荧光色素时,SYBR Green能特异性地与双链DNA结合,PCR产生的双链DNA越多,荧光信号也越强烈。利用绝对定量实时PCR,可非常灵敏地特异性地测定低至只含有单个拷贝数的DNA模板,最终定量单位为基因拷贝数/ng DNA。初始待测DNA样品的绝对浓度测定也直接影响绝对定量PCR拷贝浓度的计算精确度。相对定量PCR则用于比较两个不同的DNA样品中或不同实验条件下两个不同基因的风度和表达量,最终单位用倍数来表示。不同研究者可根据实验需要选择不同的实时PCR方法。我们先简单地介绍绝对定量PCR 。
2. PCR引物设计
假如你所研究的序列是已知的,你可以利用在线的免费程序或通过自己阅读从序列中找到合适的引物位点。Primer3 是非常好的免费在线引物设计工具。
你要做的是:
设计一对引物用于扩增该基因的较长序列,长度为 1-2kb较合适。这一PCR产物将作为绝对定量PCR的标准曲线的底物;
设计另一对引物定位于该基因的中央区域,扩增区域长度为100-200bp之间。这一PCR引物将用于定量PCR和定量RT-PCR。
定量PCR 和普通PCR引物设计的原则是一样的。我们一般遵循以下经验原则:
引物长度可选20bp
GC%为50%左右
尽量避免选择GC集中富含区
3'末端的连续GC数量不多于2
引物位点尽量为基因中央区域。
3. PCR标样的稀释制备
首先用全长引物扩增全长基因,PCR片断纯化后,用紫外分光光度计定量;
根据PCR片断的长度bp数和PCR产物浓度 ng/uL,计算出纯化后的PCR片断的拷贝浓度(copy/uL);
然后稀释PCR产物到最后5个浓度:20000,2000,200,20,2copy/uL;
从每个稀释浓度中吸取5uL加入到各个反应中;
如下图所示:从左到右的5条扩增曲线分别代表5个稀释度的PCR标样100000, 10000,1000,100,10copy/reaction;
检测到的Ct值用于标准曲线来计算未知样品中该基因的拷贝数;
利用从标样的基因拷贝数(X)和Ct值(Y),可得出Y=ALog10(X)+B,这样从未知样品的Y值可以根据公式求得该样品中的基因拷贝数X值,PCR效率Efficiency = -1+10的 (-1/A)次方。

qPCR反应过程中的荧光信号。Delta Rn为校准后的SYRB Green荧光信号,即反应过程中的荧光信号减去设备产生的基线信号。从左到右的5条扩增曲线分别代表5个稀释度的PCR标样100000, 10000,1000,100,10copy/reaction。
请联系我们
我们可提供免费的Excel表格程序,简化PCR标准样品的计算和稀释制备,您只需在表中输入PCR片断长度和PCR产物的DNA浓度就可以了。
4. 相对定量法
除了上述最简单的绝对定量法,还有相对定量法,比如ΔΔCt法。相对定量法的优势在于:1)实测中无须利用标准曲线;2)初始DNA浓度无须定量。尽管理论上如此,我们仍建议在优化过程中用标准曲线法测定PCR的效率,也确定相对定量法的可行性。在利用ΔΔCt法,要在同一RNA样品中测定两个不同基因的Ct值,一个是你所要研究的目的基因(Target gene),另一个是做为参照的内标基因(Endogenous control) ;同时,设定两个不同的采样条件或生理条件,一个是正常的生理条件作为本底参照校正值(Calibrator, or C ),另一个是你所要研究的生理条件(Unknown, or X )。这样,每个条件下可测得目的基因和内参基因的两个Ct值。相对法得到的最终值是目的基因与内参基因的比值在你所要研究的生理条件下与本底参照条件相比增长或减少的倍数,该倍数可下列公式计算得出: